El informe de investigación de la industria del “ Mercado de imágenes por resonancia magnética (MRI) ” 2021 ayuda a ofrecer la comprensión adecuada del desarrollo de la tecnología magnética. Crecimiento de la industria de imágenes por resonancia (MRI). Además, la información del mercado mundial de imágenes por resonancia magnética (IRM) en este informe permitirá establecer un estándar para los proveedores de nuevos competidores en la industria. Imágenes por resonancia magnética (IRM) El informe de mercado estimaciones principalmente para el período de 2030. En términos de datos históricos detallados, se produce un análisis profundo para el período calculado para una mejor ampliación del mercado mundial Imágenes por resonancia magnética (IRM).
“Se prevé que el mercado mundial de imágenes por resonancia magnética (IRM) aumentará a un ritmo considerable durante el período de pronóstico, entre 2021 y 2030. En 2021, el mercado está creciendo a un ritmo constante y con la creciente adopción de estrategias por parte de jugadores clave, se espera que el mercado aumente durante el horizonte proyectado “.
Este informe ha proporcionado información detallada sobre los principales impulsores de crecimiento, restricciones, desafíos, tendencias y oportunidades para ofrecer un análisis completo del mercado global Imágenes por resonancia magnética (IRM). Los participantes del mercado pueden utilizar el análisis de la dinámica del mercado para planificar estrategias de crecimiento efectivas y prepararse para los desafíos futuros de antemano.
Los principales actores en el mercado Imagen por resonancia magnética (IRM) incluyen : La investigación cubre el tamaño actual del mercado de Imágenes por resonancia magnética (IRM) del mercado y sus tasas de crecimiento basadas en registros anuales con el esquema de la compañía de jugadores / fabricantes clave:
– GE Healthcare
– Siemens
– Phillips
– Hitachi Medical Systems
– Toshiba
– Canon Medical Systems
– Tecnologías de imágenes de Aurora
– Esaote SPA
– Sanrad Medical Systems
– Imagen de aspecto
– Sistemas médicos de Neusoft
– SciMedix
– Alta tecnología de Shenzhen Anke
– Time Medical Systems
– Equipo médico de Xingaoyi
Para comprender cómo se trata el impacto de Covid-19 en este informe: https://www.precisionreports.co/enquiry/request-covid19/19291201
Análisis de segmentación:
El informe ha clasificado la industria global de imágenes por resonancia magnética (IRM) en segmentos que incluyen el tipo de producto y la aplicación. Cada segmento se evalúa en función de la tasa de crecimiento y la participación. Además, los analistas han estudiado las regiones potenciales que pueden resultar gratificantes para los fabricantes de imágenes por resonancia magnética (MRI) en los próximos años. El análisis regional incluye predicciones confiables sobre el valor y el volumen, lo que ayuda a los actores del mercado a obtener una visión profunda de la industria general de Imágenes por resonancia magnética (IRM).
Mercado dividido por tipo , este informe muestra la producción, los ingresos, el precio, la participación de mercado y la tasa de crecimiento de cada tipo, principalmente divididos en:
– Fuerza de campo baja (0-1,5 T)
– Fuerza de campo medio (1,5 T)
– Alta fuerza de campo (1. El informe de mercado de Imágenes por resonancia magnética (MRI) examina la esencia del mercado en muchas regiones del mundo. Ayuda a comprender no solo el tamaño del mercado sino también sus perspectivas de crecimiento futuro .
Segmento de mercado por región / país que incluye:
– América del Norte (Estados Unidos, Canadá y México)
– Europa (Alemania, Reino Unido, Francia, Italia, Rusia y España, etc.)
– Asia-Pacífico (China, Japón, Corea, India, Australia y el sudeste asiático, etc.)
– América del Sur (Brasil, Argentina y Colombia, etc.)
– Medio Oriente y África (Sudáfrica, Emiratos Árabes Unidos y Arabia Saudita, etc.)
Este informe proporciona un análisis histórico detallado del mercado global de imágenes por resonancia magnética (MRI) de 2015 a 2020, y proporciona amplias previsiones de mercado de 2021 a 2030 por región / país y subsectores. Cubre las ventas / ingresos / valor, el margen bruto, el crecimiento histórico y las perspectivas futuras en el mercado Imagen por resonancia magnética (IRM).
Además, el impacto de COVID-19 también está preocupado. Desde el brote en diciembre de 2019, el virus COVID-19 se ha extendido por todo el mundo y ha causado enormes pérdidas de vidas y economía, y los mercados de fabricación, turismo y financieros mundiales se han visto muy afectados, mientras que el mercado / industria en línea aumenta. Afortunadamente, con el desarrollo de vacunas y otros esfuerzos por parte de gobiernos y organizaciones globales, se espera que el impacto negativo de COVID-19 disminuya y se espera que la economía global se recupere.
Esta investigación cubre los impactos de COVID-19 en las industrias upstream, midstream y downstream. Además, esta investigación proporciona una evaluación de mercado en profundidad al resaltar información sobre varios aspectos que cubren la dinámica del mercado, como impulsores, barreras, oportunidades, amenazas y noticias y tendencias de la industria. Al final, este informe también proporciona un análisis en profundidad y consejos profesionales sobre cómo afrontar el período posterior a la COIVD-19.
Razones clave para comprar Informe de mercado de imágenes por resonancia magnética (IRM):
– El análisis del informe por geografía destacando el consumo del producto / servicio dentro de la región también indicando los factores que están afectando el mercado dentro de cada región.
– El informe brinda oportunidades y amenazas a las que se enfrentan los proveedores en la industria global de imágenes por resonancia magnética (IRM).
– El informe indica la región y el segmento que se espera que experimente el crecimiento más rápido.
– Panorama competitivo que incluye la clasificación de mercado de los principales actores, junto con el lanzamiento de nuevos productos, asociaciones, expansiones comerciales y adquisiciones.
– El informe proporciona amplios perfiles de la empresa que comprenden una descripción general de la empresa, conocimientos de la empresa, evaluación comparativa de productos y análisis FODA para los principales actores del mercado.
– El informe brinda las perspectivas de mercado presentes y futuras de la industria con respecto a los desarrollos recientes, las oportunidades de crecimiento, los impulsores, los desafíos y las restricciones de las regiones emergentes y desarrolladas.
La metodología de investigación utilizada para estimar y pronosticar este mercado comienza capturando los ingresos de los jugadores clave y sus acciones en el mercado. Se han utilizado diversas fuentes secundarias, como comunicados de prensa, informes anuales, organizaciones sin fines de lucro, asociaciones industriales, agencias gubernamentales y datos de aduanas, para identificar y recopilar información útil para este extenso estudio comercial del mercado. Los cálculos basados en esto llevaron al tamaño general del mercado. Después de llegar al tamaño general del mercado, el mercado total se ha dividido en varios segmentos y subsegmentos, que luego se han verificado a través de una investigación primaria mediante la realización de extensas entrevistas con expertos de la industria. Los procedimientos de triangulación de datos y desglose del mercado se han empleado para completar el proceso general de ingeniería del mercado y llegar a las estadísticas exactas para todos los segmentos y subsegmentos.
Algunas de las preguntas clave respondidas en este informe:
– ¿Cuál será la tasa de crecimiento del mercado, el impulso de crecimiento o la aceleración del mercado durante el período de pronóstico?
– ¿Cuáles son los factores clave que impulsan el mercado de la resonancia magnética (MRI)?
– ¿Cuál fue el tamaño del mercado emergente de imágenes por resonancia magnética (IRM) por valor en 2020?
– ¿Cuál será el tamaño del mercado emergente de imágenes por resonancia magnética (IRM) en 2030?
– ¿Qué región se espera que tenga la mayor participación de mercado en el mercado de imágenes por resonancia magnética (IRM)?
– ¿Qué tendencias, desafíos y barreras afectarán el desarrollo y el tamaño del mercado global de imágenes por resonancia magnética (MRI)?
– ¿Cuales son el volumen de ventas, los ingresos y el analisis de precios de los principales fabricantes del mercado Imagen por resonancia magnetica (IRM)?
Años considerados para este informe:
– Años históricos: 2015-2020
– Año base: 2020
– Año estimado: 2021
– Período de pronóstico del mercado de imágenes por resonancia magnética (MRI): 2021-2030
¿Por qué debería comprar este informe ?
– Proporciona conocimientos de nicho para la decisión sobre todos los segmentos posibles que ayudan en el proceso de toma de decisiones estratégicas.
– Estimación del tamaño de mercado de el mercado de imágenes por resonancia magnética (MRI) a nivel regional y global.
– Un diseño de investigación único para la estimación y el pronóstico del tamaño del mercado.
– Identificación de las principales empresas que operan en el mercado con desarrollos relacionados.
– Alcance exhaustivo para cubrir todos los segmentos posibles ayudando a todas las partes interesadas en el mercado de imágenes por resonancia magnética (MRI).
Con tablas y figuras que ayudan a analizar las tendencias mundiales del mercado Imagen de resonancia magnética (IRM) a nivel mundial, esta investigación proporciona estadísticas clave sobre el estado de la industria y es una valiosa fuente de orientación y dirección para empresas e individuos interesados en el mercado.
El informe de la industria global de imágenes por resonancia magnética (IRM) cubre los siguientes temas:
Capítulo 1 Imagen de resonancia magnética (IRM) Descripción general del mercado
1.1 Definición de imágenes por resonancia magnética (IRM)
1.2 Estado y perspectivas del tamaño del mercado de imágenes por resonancia magnética (IRM) global (2015-2030)
1.3 Comparación del tamaño del mercado mundial de imágenes por resonancia magnética (IRM) por región (2015-2030)
1.4 Comparación del tamaño del mercado mundial de imágenes por resonancia magnética (IRM) por tipo (2015-2030)
1.5 Comparación del tamaño del mercado mundial de imágenes por resonancia magnética (IRM) por aplicación (2015-2030)
1.6 Comparación del tamaño del mercado global de imágenes por resonancia magnética (IRM) por canal de ventas (2015-2030)
1.7 Dinámica del mercado de imágenes por resonancia magnética (IRM) (impactos de COVID-19)
1.7.1 Oportunidades / impulsores del mercado
1.7.2 Desafíos / riesgos del mercado
1.7.3 Noticias del mercado (Fusiones / Adquisiciones / Expansión)
1.7.4 Impactos de COVID-19 en mercado actual
1.7.5 Estrategias posteriores al brote de COVID-19
Capítulo 2 Imágenes por resonancia magnética (IRM) Análisis del segmento de mercado por jugador
2.1 Ventas globales de imágenes de resonancia magnética (MRI) y participación de mercado por jugador (2018-2020)
2.2 Ingresos y participación de mercado de las imágenes de resonancia magnética (MRI) globales por jugador (2018-2020)
2.3 Precio promedio global de resonancia magnética (MRI) por jugador (2018-2020)
2.4 Situación y tendencias de la competencia de los jugadores
2.5 Conclusión del segmento por jugador
Capítulo 3 Imágenes por resonancia magnética (IRM) Análisis del segmento de mercado por tipo
3.1 Mercado global de imágenes por resonancia magnética (IRM) por tipo
3.2 Ventas globales de imágenes de resonancia magnética (IRM) y participación de mercado por tipo (2015-2020)
3.3 Ingresos y participación de mercado de las imágenes de resonancia magnética (MRI) a nivel mundial por tipo (2015-2020)
3.4 Precio promedio global de resonancia magnética (MRI) por tipo (2015-2020)
3.5 Principales actores de la resonancia magnética (IRM) por tipo en 2020
3.6 Conclusión del segmento por tipo
Capítulo 4 Imágenes por resonancia magnética (IRM) Análisis del segmento de mercado por aplicación
4.1 Mercado global de resonancia magnética (MRI) por aplicación
4.2 Resonancia magnética global Imágenes (MRI ) Ingresos y participación de mercado por aplicación (2015 -2020)
4.3 Principales consumidores de imágenes por resonancia magnética (IRM) por aplicación en 2020
4.4 Conclusión del segmento por aplicación
Capítulo 5 Análisis de segmento de mercado de imágenes por resonancia magnética (IRM) por canal de ventas
5.1 Mercado global de imágenes por resonancia magnética (IRM) por canal de ventas
5.1.1 Canal directo
5.1.2 Canal de distribución
5.2 Ingresos y participación de mercado de las imágenes de resonancia magnética (MRI) globales por canal de ventas (2015-2020)
5.3 Principales distribuidores / distribuidores de imágenes por resonancia magnética (IRM) por canal de ventas en 2020
5.4 Conclusión del segmento por canal de ventas
Capítulo 6 Imágenes por resonancia magnética (IRM) Análisis del segmento de mercado por región
6.1 Tamaño del mercado mundial de imágenes por resonancia magnética (MRI) y CAGR por región (2015-2030)
6.2 Ventas globales de imágenes por resonancia magnética (IRM) y participación de mercado por región (2015-2020)
6.3 Ingresos y participación de mercado de las imágenes de resonancia magnética (IRM) globales por región (2015-2020)
6.4 América del Norte
6.5 Europa
6.6 Asia-Pacífico
6.7 América del Sur
6.8 Oriente Medio y África
6.9 Conclusión del segmento por región
Capítulo 7 Perfil de los principales reproductores de imágenes por resonancia magnética (IRM)
Capítulo 8 Análisis ascendente y descendente de la resonancia magnética (MRI)
8.1 Cadena industrial de imágenes por resonancia magnética (IRM)
8.2 Upstream de imágenes de resonancia magnética (MRI)
8.2.1 Materias primas
8.2.2 Coste laboral
8.2.3 Gastos de fabricación
8.2.4 Estructura de costos de fabricación
8.2.5 Proceso de fabricación
8.3 Aguas abajo de la resonancia magnética (MRI)
8.3.1 Distribuidores / distribuidores líderes de imágenes por resonancia magnética (IRM)
8.3.2 Principales consumidores de imágenes por resonancia magnética (IRM)
Capítulo 9 Tendencia de desarrollo de las imágenes por resonancia magnética (IRM) (2021-2030)
9.1 Pronóstico del tamaño del mercado global de imágenes por resonancia magnética (MRI) (ventas e ingresos) (2021-2030)
9.2 Tamaño del mercado global de resonancia magnética Imaging (MRI) y pronóstico de CAGR por región (2021-2030)
9.3 Tamaño del mercado global de imágenes por resonancia magnética (MRI) y pronóstico de CAGR por tipo (2021-2030)
9.4 Tamaño del mercado global de imágenes por resonancia magnética (IRM) y pronóstico de CAGR por aplicación (2021-2030)
9.5 Tamaño del mercado global de imágenes por resonancia magnética (MRI) y pronóstico de CAGR por canal de ventas (2021-2030)
Capítulo 10 Apéndice

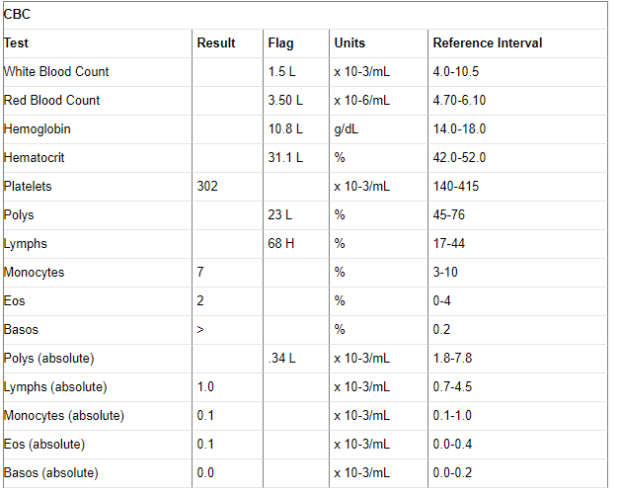
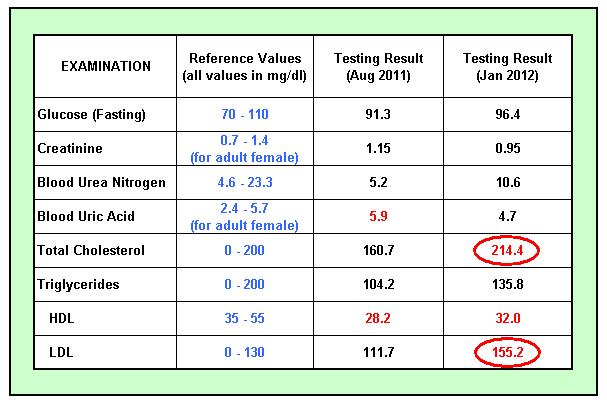 Un examen de química sanguínea básica es un procedimiento médico estándar que su médico ordenará para verificar una serie de afecciones y funciones de los órganos. Este análisis de sangre mide varias sustancias químicas en su sangre, incluidas la glucosa y la insulina. También verifica la función renal y hepática y mide otras sustancias en su cuerpo. Además de medir los niveles de estos químicos, una prueba básica también puede verificar la absorción de azúcar y el funcionamiento de sus riñones.
Un examen de química sanguínea básica es un procedimiento médico estándar que su médico ordenará para verificar una serie de afecciones y funciones de los órganos. Este análisis de sangre mide varias sustancias químicas en su sangre, incluidas la glucosa y la insulina. También verifica la función renal y hepática y mide otras sustancias en su cuerpo. Además de medir los niveles de estos químicos, una prueba básica también puede verificar la absorción de azúcar y el funcionamiento de sus riñones.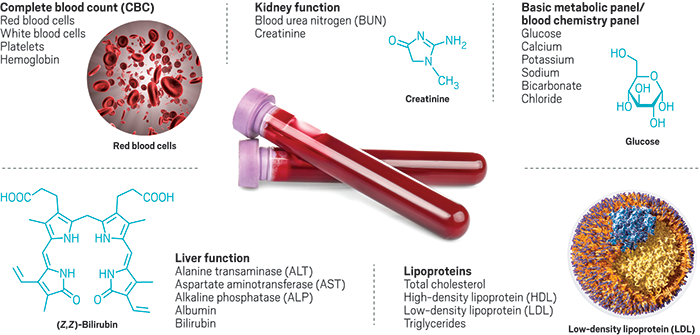 Los resultados de la prueba generalmente incluyen marcadores que indican si sus resultados se encuentran fuera del rango normal. A menudo se indican con la letra H o la letra L, o el acrónimo WNL, que significa “dentro de los límites normales”. Algunas pruebas también pueden requerir que el paciente ayune. Su proveedor de atención médica le explicará el período de ayuno y cualquier requisito adicional, si es necesario. Para un análisis de sangre rápido, es mejor ayunar durante 8 horas antes del análisis.
Los resultados de la prueba generalmente incluyen marcadores que indican si sus resultados se encuentran fuera del rango normal. A menudo se indican con la letra H o la letra L, o el acrónimo WNL, que significa “dentro de los límites normales”. Algunas pruebas también pueden requerir que el paciente ayune. Su proveedor de atención médica le explicará el período de ayuno y cualquier requisito adicional, si es necesario. Para un análisis de sangre rápido, es mejor ayunar durante 8 horas antes del análisis.
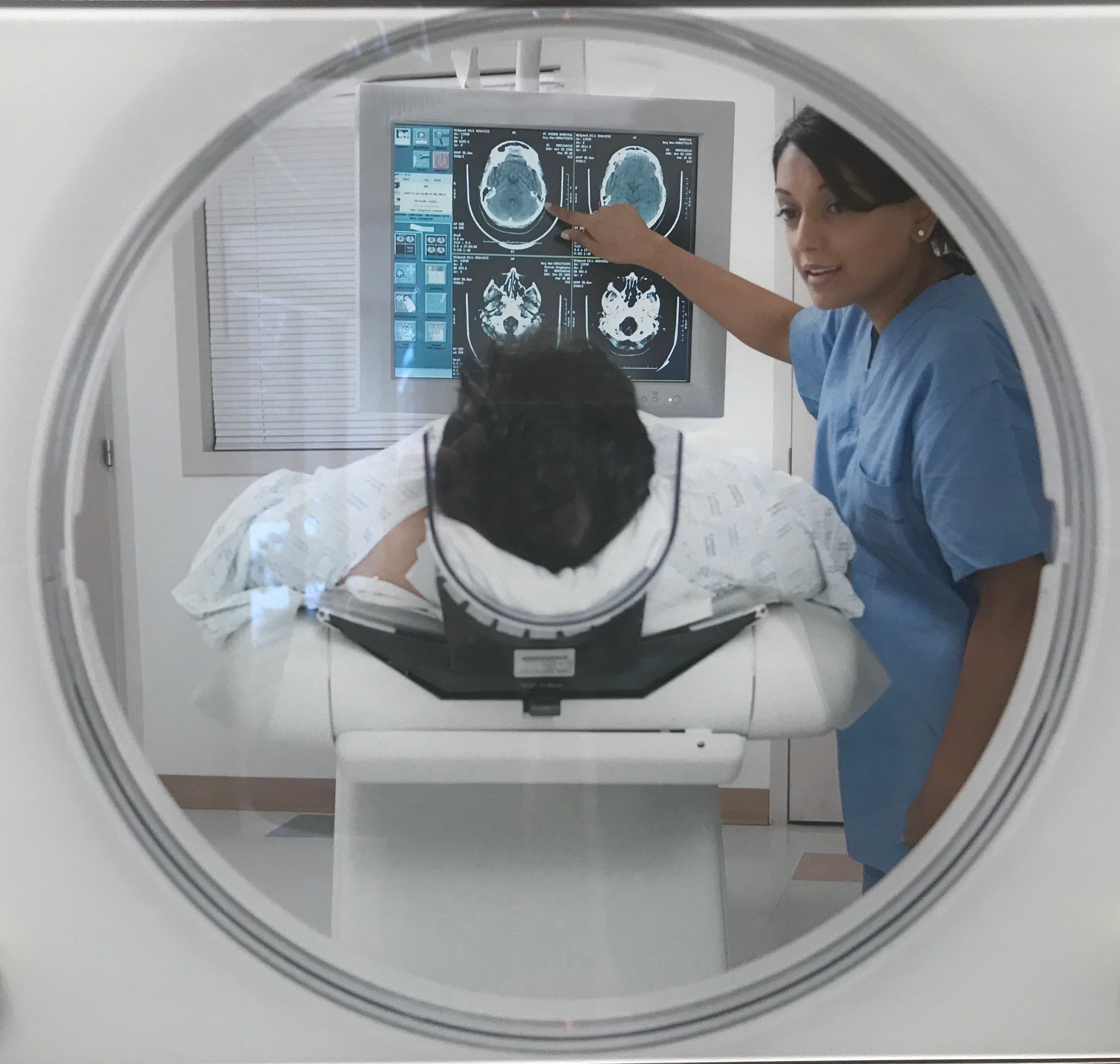 Una exploración por TEP se realiza en el departamento de medicina nuclear de un hospital o en un centro de exploración por TEP. Se le pedirá que se acueste en una mesa enganchada a una cámara y un escáner. Durante la exploración, la cámara registrará las imágenes de los órganos de su cuerpo. Luego, una computadora interpretará los resultados. Una vez que se haya sometido a la tomografía por emisión de positrones (PET), descubrirá qué debe hacer a continuación.
Una exploración por TEP se realiza en el departamento de medicina nuclear de un hospital o en un centro de exploración por TEP. Se le pedirá que se acueste en una mesa enganchada a una cámara y un escáner. Durante la exploración, la cámara registrará las imágenes de los órganos de su cuerpo. Luego, una computadora interpretará los resultados. Una vez que se haya sometido a la tomografía por emisión de positrones (PET), descubrirá qué debe hacer a continuación.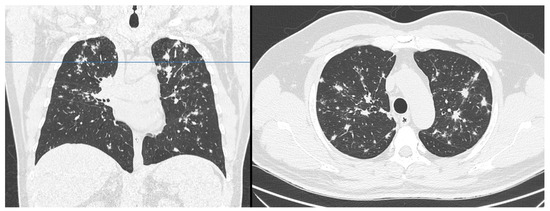


 Abby Tudor, una estudiante de cuarenta años, recientemente cumplió cuarenta. Está experimentando dolor de rodilla, dificultad para respirar al hacer ejercicio y mareos. También tiene antecedentes familiares de enfermedades cardiovasculares y ambos padres han sufrido ataques cardíacos. Sin embargo, aunque sus padres tuvieron éxito en el tratamiento de sus afecciones, Abby Tudor continuó experimentando estos síntomas a pesar de que tenía sobrepeso.
Abby Tudor, una estudiante de cuarenta años, recientemente cumplió cuarenta. Está experimentando dolor de rodilla, dificultad para respirar al hacer ejercicio y mareos. También tiene antecedentes familiares de enfermedades cardiovasculares y ambos padres han sufrido ataques cardíacos. Sin embargo, aunque sus padres tuvieron éxito en el tratamiento de sus afecciones, Abby Tudor continuó experimentando estos síntomas a pesar de que tenía sobrepeso.

 Las tomografías computarizadas son imágenes creadas midiendo la atenuación de los rayos X a medida que atraviesan un segmento del cuerpo. La diferencia entre la TC y la radiología convencional radica en el hecho de que la TC utiliza múltiples medidas para reconstruir una imagen. Estas medidas se convierten luego en unidades Hounsfield (HU), que son números enteros que se multiplican por 1000. Esto permite reconstruir imágenes con mayor precisión.
Las tomografías computarizadas son imágenes creadas midiendo la atenuación de los rayos X a medida que atraviesan un segmento del cuerpo. La diferencia entre la TC y la radiología convencional radica en el hecho de que la TC utiliza múltiples medidas para reconstruir una imagen. Estas medidas se convierten luego en unidades Hounsfield (HU), que son números enteros que se multiplican por 1000. Esto permite reconstruir imágenes con mayor precisión. Los valores de la
Los valores de la 
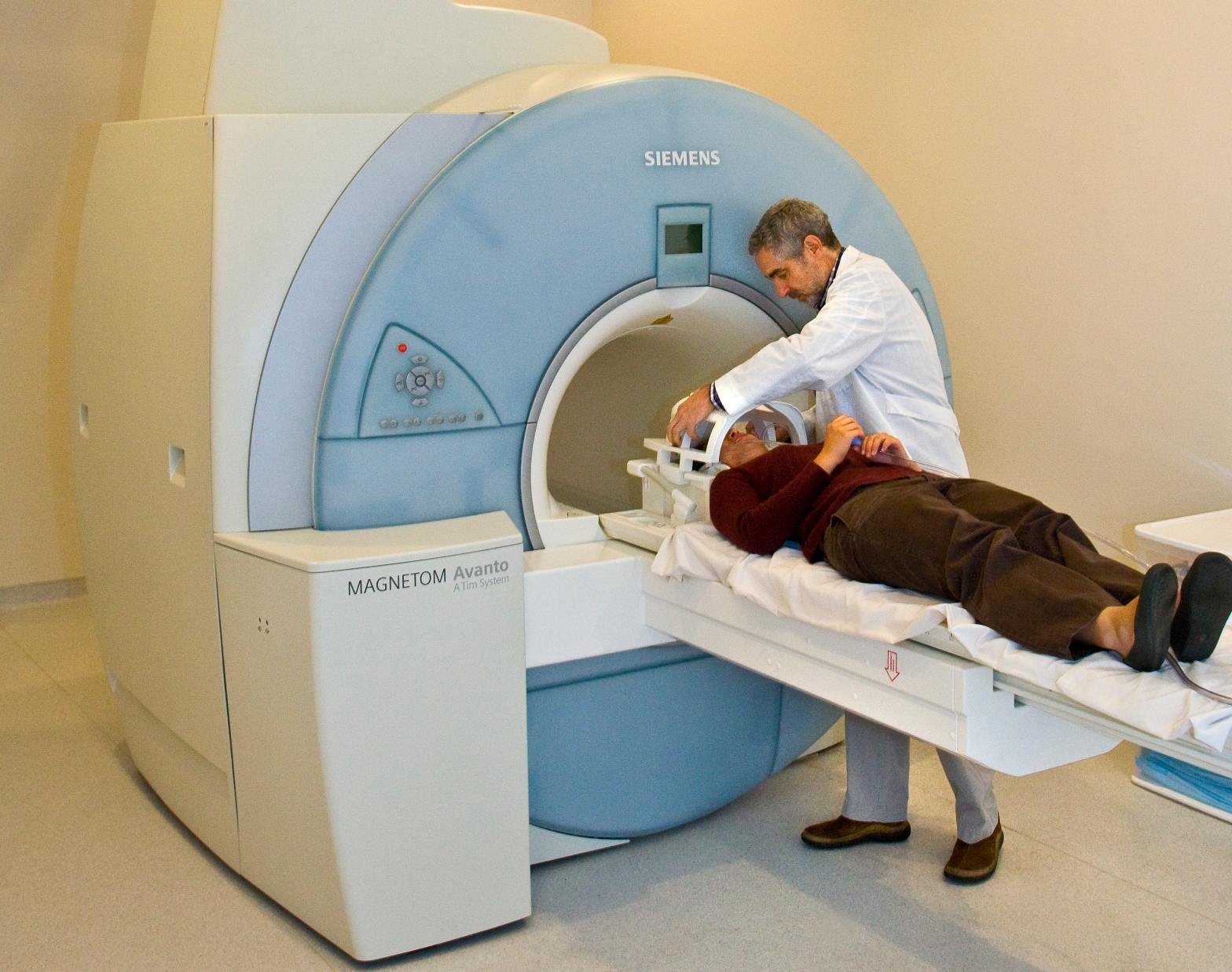

 El método del pliegue cutáneo se puede utilizar solo o con otros métodos para determinar la grasa corporal. El método es conveniente y asequible y es bastante preciso cuando se administra correctamente. Los márgenes de error rondan el 47%. Este método se basa en el supuesto de que la grasa subcutánea es proporcional a la cantidad total de peso corporal. Cuando el método se aplica correctamente, puede proporcionar una estimación bastante precisa del porcentaje de grasa corporal.
El método del pliegue cutáneo se puede utilizar solo o con otros métodos para determinar la grasa corporal. El método es conveniente y asequible y es bastante preciso cuando se administra correctamente. Los márgenes de error rondan el 47%. Este método se basa en el supuesto de que la grasa subcutánea es proporcional a la cantidad total de peso corporal. Cuando el método se aplica correctamente, puede proporcionar una estimación bastante precisa del porcentaje de grasa corporal.